Diagnose
Eine PAH ist schwierig zu erkennen, da die Symptome anderen Lungenerkrankungen wie Asthma und COPD sehr ähnlich sind. Deshalb werden im Rahmen der Diagnosestellung zunächst andere, häufiger vorkommende Krankheiten ausgeschlossen und während des Diagnoseverfahrens Lungen- und Herzspezialist:innen hinzugezogen.
Häufig vergehen zwischen dem Auftreten der ersten Symptome und der PAH-Diagnose mehr als 2 Jahre.
Unter Diagnose der pulmonalen Hypertonie sind die verschiedenen Diagnoseuntersuchungen ausführlich beschrieben.
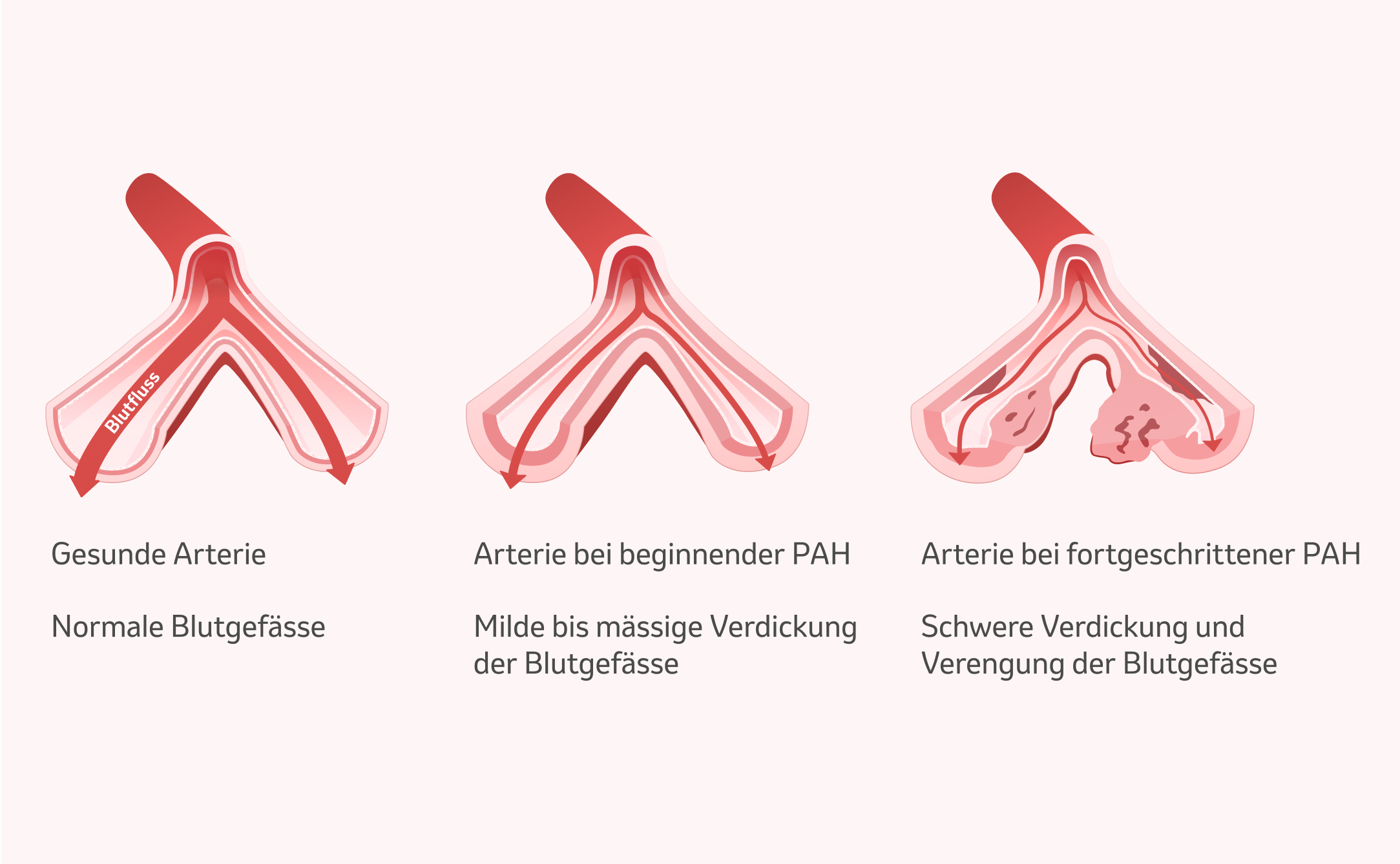
Einteilung der PAH gemäss WHO-Klassifikation
Die PAH ist die erste von fünf Gruppen der pulmonalen Hypertonie, die von der WHO definiert wurden. Nach ihren Ursachen wird die pulmonale arterielle Hypertonie wie folgt unterschieden:
1. Pulmonale arterielle Hypertonie (PAH)
1.1 Idiopathische PAH
Die idiopathische tritt ohne eindeutige Ursache auf.
1.2 Hereditäre PAH
Die vererbbare PAH steht in Zusammenhang mit Genen, die von Familienmitgliedern weitergegeben werden.
1.3 PAH ausgelöst durch bestimmte Medikamente oder Toxine
Eine PAH kann auch aufgrund von bestimmten Medikamenten oder Drogen ausgelöst werden. Dazu zählen bestimmte Medikamente zur Gewichtsabnahme, die unterdessen nicht mehr auf dem Markt sind, sowie bestimmte Antidepressiva. Kokain und einige Chemotherapien gelten als mögliche Risikofaktoren, Amphetamine und Methamphetamine, Substanzen mit einer stark stimulierenden Wirkung auf das zentrale Nervensystem, gelten als wahrscheinliche Risikofaktoren für die Entstehung einer PAH.
1.4 PAH in Verbindung mit anderen Erkrankungen
PAH kann sich auch in Verbindung mit anderen Erkrankungen entwickeln, wie z.B.
- Bindegewebserkrankungen
- HIV-Infektion
- portale Hypertonie
- angeborene Herzfehler
- Erkrankungen mit vorwiegender Beteiligung der Lungenvenen
- Schistosomiasis (Wurmerkrankung).
1.5 PAH mit Merkmalen einer venösen/kapillaren (PVOD/ PCH) Beteiligung
Bei dieser Krankheitsform sind neben den Arterien auch die Venen und Kapillaren der Lunge betroffen.
1.6 Persistierende PAH des Neugeborenen
Bei dieser Form der PAH bleibt der beim Kind im Mutterleib natürlich erhöhte Lungengefässdruck auch nach der Geburt bestehen.
Unter Formen der pulmonalen Hypertonie sind die fünf Gruppen der pulmonalen Hypertonie gemäss WHO-Klassifikation näher beschrieben.